Menopausal transition
- Ann Intern Med. 2009 Apr 7;150(7):ITC4-1-15
- Best Pract Res Clin Obstet Gynaecol. 2009 Feb;23(1):25-32.
- Med Clin North Am. 2008 Sep;92(5):1253-71, xii.
- Williams Gynecology, ch. 16, 21
Definitions and overview
- Menopause: point in time 1 year after the cessation of menstruation (retroactive definition)
- Menopausal transition: the time period from menstrual cycle irregularity to final menstrual period
- Perimenopause: menopausal transition + 1 year following final menstrual period
- Postmenopause: all years following final menstrual period
- Premature ovarian failure (also called premature ovarian insufficiency): cessation of menses prior to age of 40
- Median age at menopause is 52; preserved across cultures and races
- Certain factors hasten menopause: smoking, high fiber or vegetarian diet, low body mass index, type I diabetes mellitus and no previous pregnancies (nulligravida).
|
Stage terminology |
STRAW stage |
Menstrual cycle |
FSH level |
LH level |
Menopause symptom prevalence |
|
Early reproductive |
-5 |
variable → regular |
normal |
normal |
none |
|
Peak reproductive |
-4 |
regular |
normal |
normal |
none |
|
Late reproductive |
-3 |
regular |
↑ |
normal to ↑ |
sporadic |
|
Early menopausal transition |
-2 |
variable length (>7days from normal) |
↑↑ |
↑↑ |
vasomotor 15-20% vaginal — |
|
Late menopausal transition |
-1 |
≥ 2 skipped cycles + interval of amenorrhea of ≥ 60 days |
↑↑ |
↑↑ |
vasomotor 20-30% vaginal — |
|
Final menstrual period (FMP) |
0 |
|
|||
|
Early postmenopause |
+1a = menopause +1b = years 2-5 after FMP |
+1a = 12 mos amenorrhea +1b = none |
↑↑↑ |
↑↑↑ |
vasomotor 35-55% vaginal 10-30% |
|
Late postmenopause |
+2 |
none |
↑↑↑ |
↑↑↑ |
vasomotor 30%& declining vaginal 35-47% |
STRAW: Stages of Reproductive Aging Workshop
The hypothalamic pituitary gonadal axis
- Please see section on Menstrual cycle chapter for premenopausal physiology
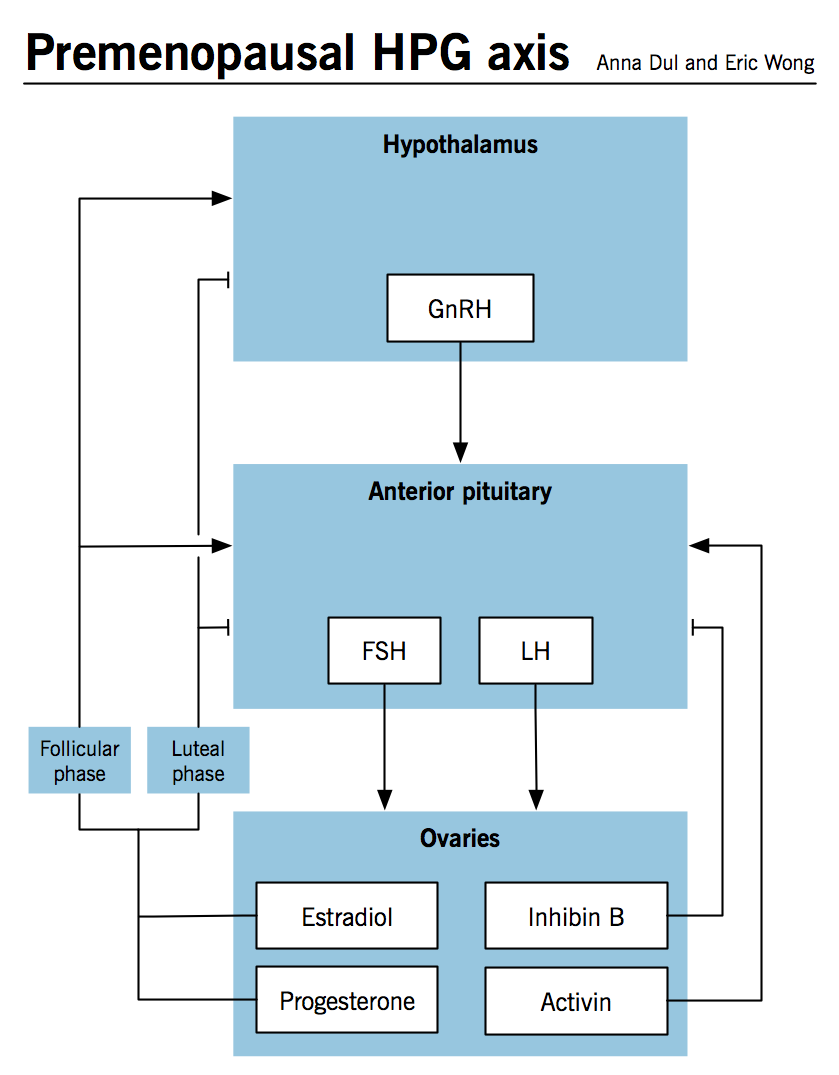
- Ovarian senescence begins in utero and progresses with primordial follicle activation, maturation and regression
- Follicular phase of menstrual cycle decreases with aging (first seen during late reproductive stage)
- Menstrual cycles become irregular during early menopausal transition because of fluctuant anterior pituitary gonadotropins
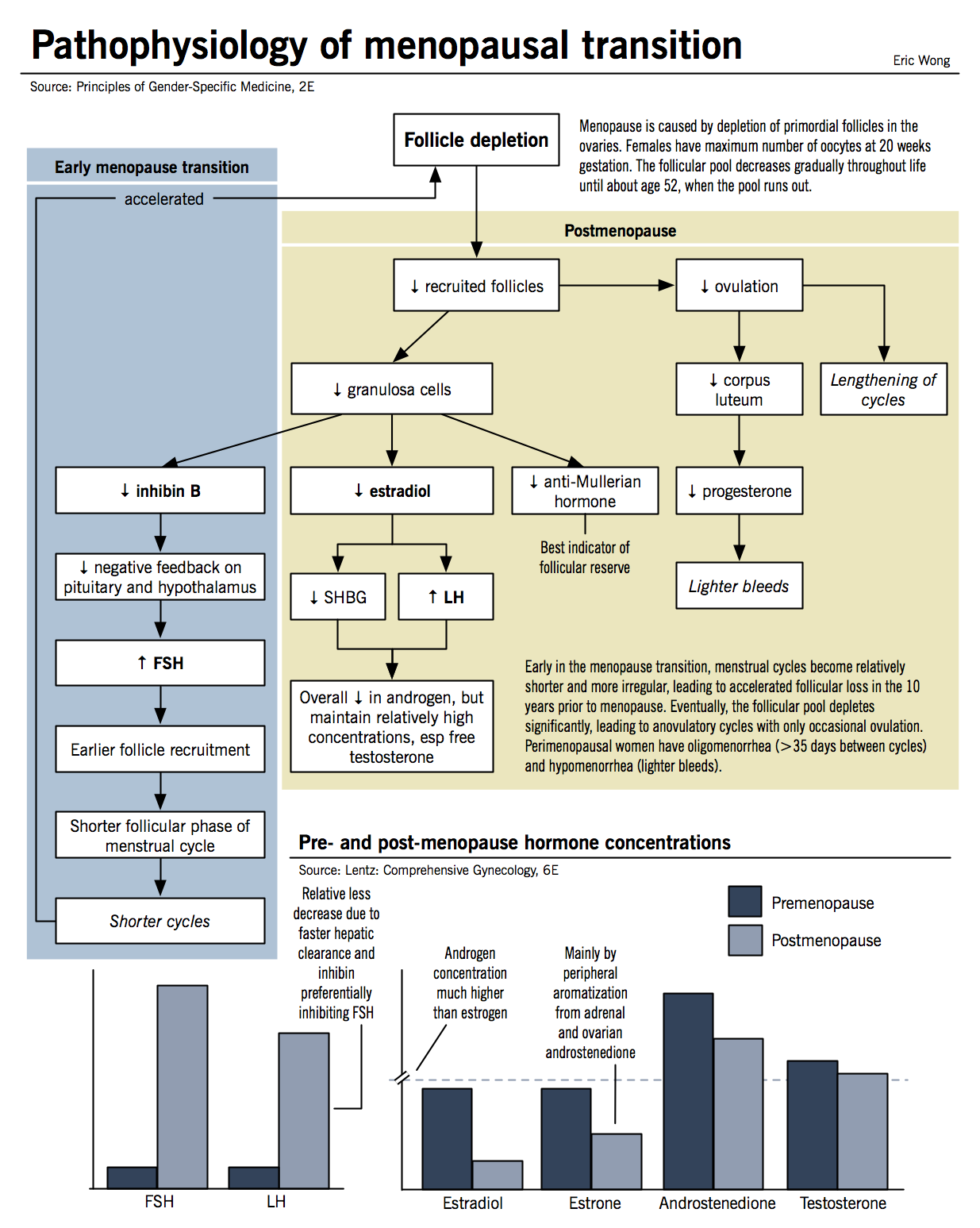
- Decreased inhibin B is the primary initiator of menopause
- Decreases in early menopausal transition
- Normal function is to negatively inhibit the anterior pituitary from secreting FSH early in the menstrual cycle
- As the female ages, the levels of inhibin B decrease due to declining/poorly functioning follicle numbers → decreased inhibin B causes increased early cycle FSH because the negative inhibition has been removed
- Ovaries respond to increased FSH by secreting estradiol because the FSH has increased the number of recruited follicles in each cohort → overall number of follicles decreased but estradiol levels normal/high due to high serum FSH and increase in aromatase activity
- Aromatase converts testosterone to estradiol
- Progesterone further decreases during the luteal phase of the cycle after inhibin B levels decrease
- When all follicles are depleted the ovary becomes unresponsive to vastly increased FSH and estradiol levels then decrease
- LH continues to stimulate the secretion of androgens
Pathophysiology and clinical features
- Classic symptoms of menopause are well known colloquially but there are also important systemic effects that impact wellness taking place during menopausal transition into the post-menopausal period
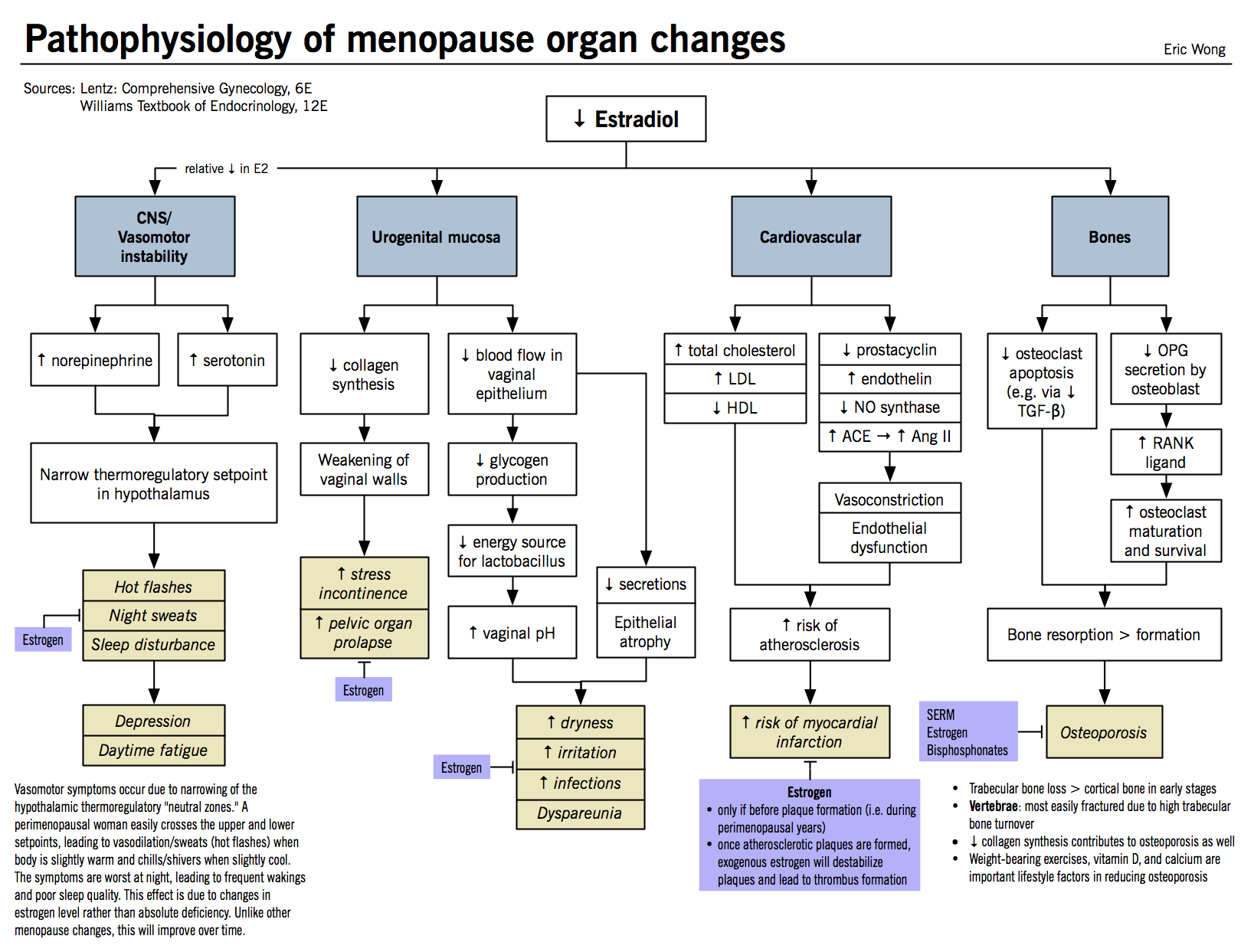
Cardiovascular disease
- Estrogen is cardioprotective because it increases high density lipoprotein levels and decreases low density lipoprotein levels
- Post-menopausal women lack estrogen and their risk of developing cardiovascular disease catches up to that of a male counterpart at the age of 70
- Truncal obesity, an independent risk factor of cardiovascular disease, is controlled by the relative proportion of androgens
- High androgen state is a risk factor → decreased estrogens + insulin resistance can contribute to the state
- Risk can be managed by the normal preventative strategies; use of hormone replacement is not indicated
Ovarian change
- As noted above, programmed ovarian senescence is accelerated as the woman ages
- Post-menopausal women have marked anatomical differences due to the senescence, including: smaller volume, lack of follicular cysts, increased atretic follicles, and persistent corpora albicans
Endometrial change
- Endometrium responds to serum estrogen and progesterone levels, which fluctuate depending on stage of menopausal transition
- Early: fluctuant endometrial thickening in response to estrogen levels and opposed by progesterone
- Later: anovulation → estrogen no longer opposed by progesterone → increased proliferation and disorganization of endometrial tissue
- Estrogen derived from extragonadal aromatization of androgen to estrogen because of obesity
- Decreased sex-hormone binding globulin (SHBG) increases the levels of free, bioavailable estrogen
- Post-menopause: no estrogen → atrophy and cystic changes
Urogenital tract change
- Estrogen and progesterone receptors throughout the tract
- Lack of substrate leads to: decreased vascularity → decreased muscle, decreased epithelial lining, increased fatty deposits → irritation, burning, itching and lack of lubrication
- Dyspareunia from the above changes and subsequent vaginal atrophy leading to increased tissue trauma and bleeding
- Less glycogen and lactic acid → pH changes to a more alkaline state (from ~4.5 to ~7) → increased susceptibility to infection
- Receptors on connective tissue can affect the laxity of ligamentous structures of the pelvic floor → incontinence and prolapse of pelvic structures into the vagina increases after menopause
Breast changes
- Ductal structures affected by estrogen
- Lobular structures affected by progesterone
- Decreased serum levels of both in the postmenopausal period leads to loss of volume
- Replacement of dense breast tissue with soft adipose
Vasomotor symptoms
Mayo Clin Proc. 2002 Nov;77(11):1207-18.
- Clinical features:
- Most common in the late menopausal transition and early postmenopause, when fluctuation in estrogen level is greatest
- Characterized by sudden-onset hot flashes/flushes and sweats, followed by chills
- Usually initiates in upper torso; lasts 1-5 minutes; more common at night
- Triggered by stress, warm environment, spicy foods
- Pathophysiology:
- Normally, the body withstands a reasonable range of core temperatures without triggering heat dissipation mechanisms. The range of core temperatures is called the thermoneutral zone, and it is controlled by the thermoregulatory center in the medial preoptic area of the hypothalamus.
- Rapid decreases in estrogen levels (estrogen withdrawal) causes:
- ↑ norepinephrine stimulation
- ↑ serotonin activation
- ↑ activity of central opioid peptides
- These factors added together causes narrowing of the thermoneutral zone in the thermoregulatory center.
- The upper threshold is decreased, which means smaller increases in core body temperature can trigger mechanisms to dissipate heat, such as vasodilation and sweating
- The lower threshold is increased, which means smaller decreases in core temperature can trigger mechanisms to preserve heat, such as shivers and chills
- Hot flashes start with core temperature rising above the narrow upper threshold → vasodilation in the peripheral vasculature → ↑ skin temperature, ↑ systolic BP, ↑ heart rate
- Can be accompanied by anxiety and palpitations
- Reflex cooling from the improved skin surface heat dissipation causes chills after the flash
- Commonly occur at night, which leads to sleep dysfunction and reports of fatigue because of disruptive symptoms
- Estrogens effective for treatment of vasomotor symptoms
Bone changes
- Peak bone mass at age of 25-35 after which it declines steadily
- Bone remodelling is done by osteoclasts (destruction of bone matrix) and osteoblasts (building of lamellar bone)
- Osteocytes = osteoblasts trapped in the matrix
- RANK and RANKL interactions promote the differentiation of osteoclast precursors into mature osteoclasts
- OPG can bind to RANKL and interrupt this process
- Estrogen stimulates secretion of OPG by the osteoblast
- More factors at play but this is a simplification of the pathway related to menopause
- Osteoporosis typically affects trabecular bone → defect in bone mineral density
- Menopause causes primary osteoporosis: estrogen decrease → increased osteoclast formation → bone resorption > bone formation
- Loss of estrogen also sensitizes bone to respond highly to parathyroid hormone
- When serum calcium becomes low, the parathyroid releases PTH to stimulate vitamin D production
- Vitamin D increases absorption of calcium in the kidney and the intestine as well as stimulating osteoclasts
- With no estrogen, the bone now releases more calcium for the same amount of PTH stimulation further weakening the structure
- When serum calcium becomes low, the parathyroid releases PTH to stimulate vitamin D production
- Dental loss because of osteoporosis of the alveolar bone and decreased salivary secretions which protect against dental caries
Skin changes
- Difficult to differentiate from photoexposure changes
- Thinning and loss of elasticity due to decreased collagen content, dry due to decreased sebaceous gland secretion and decreased vascularity.
Psychological changes
- Controversy regarding the validity of claims that menopausal transition affects cognition and/or mood
- Leading thought centers on idea that high and erratic estradiol levels relate to emotional distress
- Perceived symptoms may be a result of emotional distress over menopausal symptoms as fluctuant estrogen levels are correlated with menopausal symptom intensity
- Leading thought centers on idea that high and erratic estradiol levels relate to emotional distress
Premature ovarian insufficiency (POI)
- Lancet. 2010 Sep 11;376(9744):911-21.
- Obstet Gynecol. 2009 Jun;113(6):1355-63.
- Reproduction. 2010 Nov;140(5):633-41.
- Commonly referred to as premature ovarian failure (POF) but this is a misnomer as there is varying ovarian function in ~50% of patients with POI
- Formal diagnoses made via history of amenorrhea for >4 months + 2 serum FSH levels gathered >1 month apart in a woman
- POI is ovarian failure with a high FSH level (primary hypogonadism)
- Loss of oocytes → lack of folliculogenesis → lack of ovarian estrogen production
- Clinical presentation is as per menopause when at the full end of the spectrum of insufficiency → amenorrhea and estrogen deficiency symptoms
|
Spectrum of POI |
FSH |
Menstruation |
Reproductive capacity |
|
Normal |
Normal |
Normal |
Regular |
|
Occult |
Normal |
↓ |
Regular |
|
Biochemical |
↑ |
↓ |
Regular |
|
Overt |
↑ |
↓ |
Intermittent → absent |
Etiology and pathogenesis
- Two broad categories:
- Follicle depletion → lack of follicles due to inadequate initial pool of primordial follicles, increased expenditure, or destruction because of a follicle targeting toxin
- Follicle dysfunction → follicles present but non-functional in steroid production due to a pathological mechanism
Follicle depletion
- Depletion due to genetics (X chromosome disorders or somatic chromosome mutations), autoimmune cause, and ovarian toxins
X chromosome disorders
Turner syndrome
- Genotype is XO; can be a mosaicism with expected range of clinical presentation
- Turner fetuses contain normal number of primordial follicle germ cells up to week 12 of gestation with subsequent decrease in number → therefore it is accelerated atresia during the gestational period
- Ovaries described as streak gonads with extensive connective tissue infiltration and absent to few atretic follicles
Fragile X syndrome premutation
- X-linked form of severe intellectual disability affecting the 5’ untranslated portion of the FMR1 gene
- Full mutation (affected): >200 CGG repeats in the FMR1 region
- No POI
- Premutation: 55 → 200 CGG repeats
- Can expand to a full mutation in one generation of transmission
- Associated with POI → increasing number of repeats decreases age at menopause
- Gray zone: 40 → 55 CGG repeats
- Can be associated with POI
- Normal:
- Full mutation (affected): >200 CGG repeats in the FMR1 region
- When a female passes on her premutation it can increase the number of CGG repeats and develop into a full mutation, whereas in males the mutation can regress, expand (shy of a full mutation), or stay the same
- Mechanism for initiation of POI in premutations of Fragile X syndrome is unknown
- Potentially increased FMR1 mRNA expression and decreased FMR1 protein
- Full mutation may not have an effect on ovarian failure because they lack ability to produce FMR1 protein → FMR1 mRNA proposed as the toxic effect
- Follicular phase reduced
- Mean follicular phase FSH increased
- Inhibin B (follicular phase), inhibin A/progesterone (luteal phase) reduced
- Potentially increased FMR1 mRNA expression and decreased FMR1 protein
Other X chromosomal mutations
- Any mutations affecting portions of the X chromosome essential for ovarian development/function can cause POI
- Regions from Xq13-Xq21 and Xq23-Xq26, and DIAPH2 important locations
Somatic chromosome mutations
Galactosemia
- Galactose metabolites are toxic to the ovaries and cause hypogonadism → genetic mutations affecting the coding portion of genes involved in galactose metabolism will cause an accumulation of metabolites
Bone morphogenetic protein-15
- BMP-15 is a tumor growth factor that regulates ovulation and folliculogenesis
- Causes reduced growth of granulosa cells
Miscellaneous somatic mutations
- Various gene mutations have been implicated:
- FSH receptor mutations
- BPES type I → mutation of FOXL2 gene
Autoimmune causes
- Usually part of a polyglandular autoimmune failure (associated with other endocrine failures)
- Possible exposure to a virus may be a trigger to develop autoimmunity → molecular mimicry of activated lymphocytes/antibodies may begin to target ovarian tissue
- POI + adrenal autoimmune disease → infiltration of thecal cells, primordial follicle unaffected
Ovarian toxins
- Direct destructive effects on ovarian tissues
- Chemotherapy most common culprit, especially alkylating agents
- Infective causes:
- Mumps virus has been suspected to cause inflammatory oophoritis, which leads to POI
- Cytomegalovirus – only from case reports
Follicle dysfunction
- Mutations to intraovarian modulators, steroidogenic enzymes or receptors to gonadotropins
- Disorders that affect the production of estradiol precursors or the conversion of testosterone to estradiol (aromatase function) → decreased estradiol and no FSH negative feedback
Intraovarian modulator abnormalities
- Paracrine regulators can decrease the response in the ovaries to FSH and LH or directly impact estrogen/progesterone production
- Examples: abnormalities to α-subunit of inhibin, bone morphogenetic protein (mechanism as under follicular depletion described above)
Gonadotropin receptor abnormalities
- Mutations that prevent the binding of FSH or cannot communicate the signal from bound FSH intracellularly → estrogen deficiency because of lack of signal despite increasing gonadotropin
- Can also affect LH receptors and signalling → effect on progesterone
Steroidogenic enzyme abnormalities
- Low estrogen, high FSH levels in subtypes of congenital adrenal hyperplasia
- Steroidogenic acute regulatory enzyme (StAR) → ineffective transport of cholesterol to mitochondrial membrane important for estradiol synthesis
- CYP17 mutations → CYP17 an important catalyst in production of progestin, estrogens and androgens (among other hormones)