Definition
Deep vein thrombosis (DVT) and pulmonary embolism (PE) are manifestations of the same pathological entity, called venous thromboembolism (VTE).
- An embolus is any intravascular material that migrates from its original location to occlude a distal vessel. Although the embolus can be a blood clot (thrombus), fat, air, amniotic fluid, or tumour, a PE is usually caused by a thrombus originating from the deep veins in the legs (deep venous thrombosis, DVT).
Arterial vs. venous thrombosis
- Thromb Haemost. 2011 Apr;105(4):586-96.
The coagulation cascade is an essential part of hemostasis. However, the same coagulation factors can give rise to clot formation in the circulation that is inappropriate (i.e. not for hemostasis). Thrombi can form in both the arteries and veins, but they have different pathophysiology and lead to different outcomes. This chapter is about venous thrombosis.
|
|
Arterial thrombosis |
Venous thrombosis (VTE) |
|
Mechanism |
Typically from rupture of atherosclerotic plaques. |
Typically from a combination of factors from Virchow’s triad. |
|
Location |
Left heart chambers, arteries |
Venous sinusoids of muscles and valves in veins |
|
Diseases |
Acute coronary syndrome Ischemic stroke Limb claudication/ischemia |
Deep venous thrombosis Pulmonary embolism |
|
Composition |
Mainly platelets |
Mainly fibrin |
|
Treatment |
Mainly antiplatelet agents (ASA, clopidogrel) |
Mainly anticoagulants (heparins, warfarin) |
Etiology
- N Engl J Med. 2008 Mar 6;358(10):1037-52.
- J Cardiovasc Nurs. 2005 Jul-Aug;20(4):254-9.
Venous thromboembolism is associated with Virchow’s triad: three conditions that predispose to thrombus formation.
- Hypercoagulability
- Stasis
- Endothelial damage
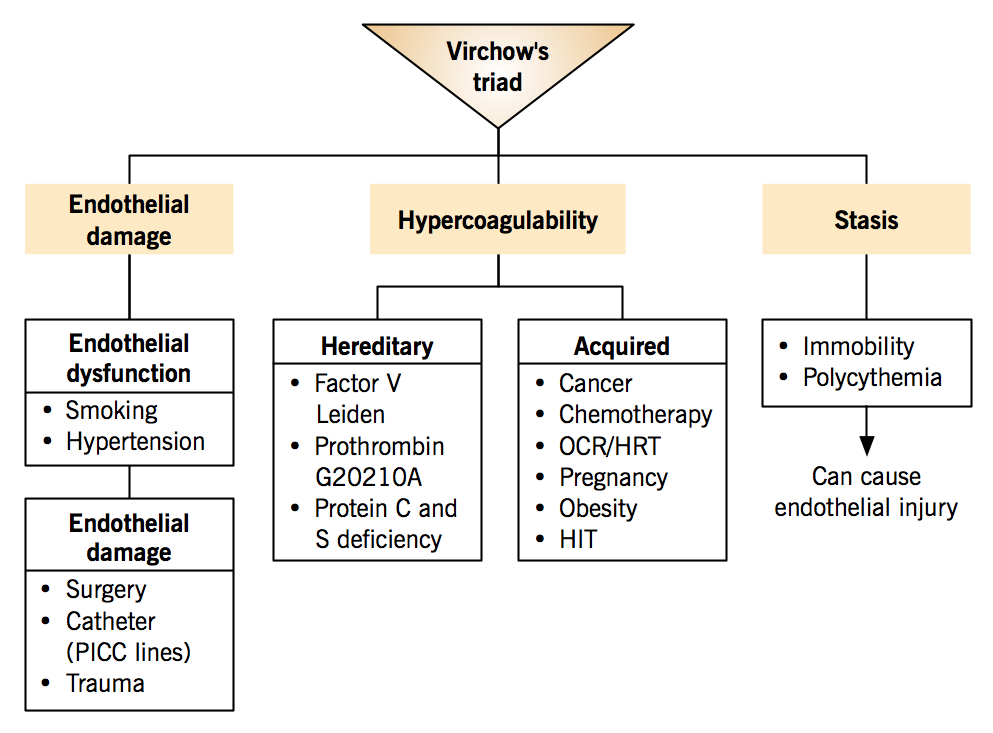
VTE often arise from the synergistic effects of multiple risk factors, for example, when a patient with inherited factor V Leiden mutation uses oral contraceptives (acquired risk on genetic risk background).
| Triad component | Associated risk factors |
|
Hypercoagulability
Changes in blood coagulation pathway, shifting balance toward coagulation
|
Hereditary factors (inherited thrombophilia)
*The 2 most common hereditary factors; autosomal dominant risk inheritance Acquired factors
|
|
Stasis
The slowing or stopping of blood flow
|
|
| Endothelial damage Normal endothelium is antithrombotic. |
Endothelial dysfunction: Shifts the balance between clot generation and breakdown towards thrombosis due to decreased synthesis of nitric oxide and prostacyclin and increased endothelin-1.
Endothelial damage: Exposure of subendothelial tissue factor and collagen, which offer a substrate for platelet binding, activation and aggregation; leading to clot formation.
|
Pathophysiology of DVT
- Semin Nucl Med. 2001 Apr;31(2):90-101.
- Crit Care Clin. 2011 Oct;27(4):869-84, vi.
- Circulation. 2003 Jun 17;107(23 Suppl 1):I22-30.
Deep venous thrombosis usually arises in the lower extremities. Most DVTs form in the calf veins, particularly in the soleus sinusoids and cusps of the valves.
- Venous valves are avascular, which, in conjunction with reduced flow of oxygenated blood in veins, predisposes the endothelium to be hypoxemic. The endothelium around valves responds by expressing adhesion molecules that attract leukocytes. These cells transfer tissue factor to the endothelium, which can complex with activated factor VII to begin the coagulation cascade via the extrinsic pathway. The main component of these venous thrombi is fibrin (as product of coagulation cascade) and red blood cells, which get trapped in the clot. Platelets also contribute, but to a lesser extent.
- The skeletal muscle pump helps prevent DVT by moving blood past the valves (i.e. reducing venous stasis), which washes away activated clotting factors that can otherwise propagate the initial thrombus.
- If a clot forms and does not resolve (see below), it will extend proximally into the popliteal and femoral veins (“proximal veins”). 25% of calf DVTs will extend proximally within 7 days. While calf DVTs are usually asymptomatic and do not give rise to significant PEs, proximal DVTs are more likely symptomatic and can embolize to form dangerous PEs.
By the numbers
- 96% arise in the lower extremities; 4% arise in the upper extremities.
Chest. 2008 Jan;133(1):143-8. - Of symptomatic lower-extremity DVTs, 88% involve the proximal veins; the rest only involve the calf veins. Almost all lower-extremity DVTs arise from the calf veins and extend proximally.
Arch Intern Med. 1993 Dec 27;153(24):2777-80. - 90% of PEs arise from DVTs.
- 50% of symptomatic proximal lower-extremity DVTs have asymptomatic PEs.
- 70% of PEs have asymptomatic DVTs.
- 28% of symptomatic DVTs will have post-thrombotic syndrome after 5 years.
Resolution and consequences
The initial thrombus can lead to complete resolution, clot extension/embolization, or organization.
- Complete resolution: Fibrinolysis is a dynamic process where plasminogen is converted into plasmin, an enzyme that degrades fibrin into soluble peptides. Fibrinolysis starts within hours, and it can lead to complete or partial resolution of the thrombus. Partial resolution may lead to any one of these 3 consequences.
- Clot extension and embolization: Proximal flow of the venous blood sweeps the thrombus in the same direction, extending it into the proximal veins.
- Organization: Thrombi that do not resolve begin to retract within days. At the same time, inflammatory cells infiltrate the thrombi and cause remodeling. The residual clot is incorporated into the vessel wall and a layer of endothelial cells forms on top (re-endothelialization). This process, called organization, allows some blood flow to resume, but it destroys valves along the length of the clot and causes scarring of the veins. The hemodynamic changes to the vein causes post-thrombotic syndrome.
- Post-thrombotic syndrome is a consequence of DVTs, and the clinical features include pain, leg edema, and other signs of venous insufficiency. It occurs in approximately 1/3 of DVT cases. The cause is a combination of venous obstruction by residual clots or venous scarring and venous reflux due to valve destruction. Prevention of this sequela includes adequate anticoagulation to prevent VTE recurrence and compression stockings to improve venous return.
Pathophysiology of PE
Hellenic J Cardiol. 2007 Mar-Apr;48(2):94-107.
Circulation. 2003 Dec 2;108(22):2726-9.
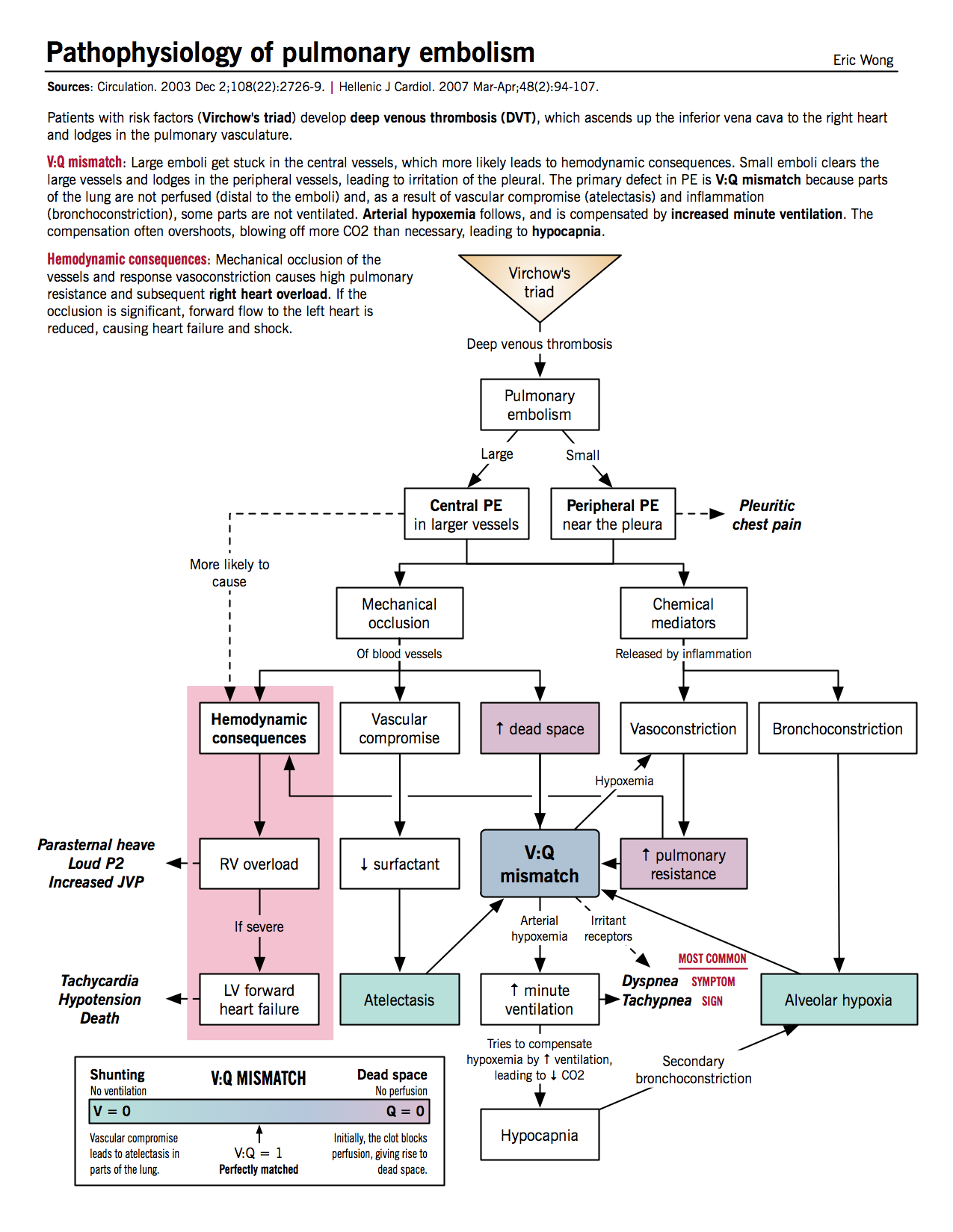
Effects of mechanical occlusion
- Increased alveolar (physiologic) dead space: decreased perfusion of alveoli distal to thrombus causes the alveoli to be ventilated but not perfused, resulting in V/Q mismatch (high V/Q) and increased dead space
- Increased minute ventilation: patient compensates for dead space and responds to chemical irritation by hyperventilation.
- Hypocapnia: increased minute ventilation causes decreased blood CO2 and respiratory alkalosis. Hypocapnia exacerbates alveolar hypoxemia by causing secondary bronchoconstriction.
- Increased pulmonary vascular resistance: due to vascular obstruction by thrombus and chemical mediators from platelets (see below)
- Decreased surfactant and atelectasis: vascular compromise beyond thrombus reduces surfactant production and thus predisposes distal region to atelectasis
Effects of chemical mediators
- Platelets from the thrombus secrete chemical mediators such as histamine and serotonin, which causes pulmonary vasoconstriction and bronchoconstriction.
- Bronchoconstriction leads to alveolar hypoxemia, which in turn causes more vasoconstriction and increased vascular resistance.
Hemodynamic consequences
- Increased right ventricular afterload: from increased pulmonary vascular resistance.
- Right ventricular dilatation and hypertrophy: parasternal heave, loud P2
- RV ischemia
- Right-sided (backward) heart failure: increased jugular venous pressure (JVP)
- Right ventricular dilatation and hypertrophy: parasternal heave, loud P2
- Decreased left ventricular filling: because of bowing of interventricular septum to left side from RV hypertrophy
- Left-sided (forward) heart failure: hypotension, syncope, cardiogenic shock
Resolution
- Intrinsic thrombolytic mechanisms (plasmin) start to lyse clots: D-dimer (breakdown product of fibrin) levels increase in serum.
- Symptomatic PE is treated with anticoagulation therapy (oral or parenteral), thrombolytic therapy (for massive PE causing cardiogenic shock), or inferior vena cava filter (if anticoagulation is contraindicated). See Treatment section for details.
- Untreated large PE causes death by acute increase in right ventricular pressure, leading to RV failure.
Clinical features of DVT
- Semin Nucl Med. 2001 Apr;31(2):90-101.
- JAMA. 1998 Apr 8;279(14):1094-9.
Clinicians accurately diagnose DVT using clinical features in approximately 25% of cases because the signs and symptoms are neither sensitive nor specific. Therefore, it is important to confirm clinical findings using additional testing, such as compression ultrasonography. The signs and symptoms of DVT arise from (i) venous obstruction and (ii) inflammation of the veins. Patients may also present with features of pulmonary embolism.
|
Symptoms |
Signs |
Mechanism |
|
Asymmetric leg/calf swelling |
Pitting edema on affect side |
Swelling and pitting edema are caused by venous obstruction. Calf circumference is measured 10cm below the tibial tuberosity. Normal difference between the two legs should be less than 1cm; greater than 3cm difference is considered significant. |
|
Pain, erythema |
Localized tenderness along deep venous system |
Pain, erythema, and tenderness are caused by vascular inflammation. Recruitment of inflammatory cells to thrombus and venous stasis causes phlebitis. |
|
|
Homans sign |
First observed by surgeon Dr. John Homans, the sign is elicited by passive dorsiflexion of the ankle. Positive findings include increased resistance to dorsiflexion or knee flexion, both in response to irritation of the posterior calf muscles. This sign is neither sensitive nor specific. N Engl J Med. 1946 Aug 1;235(5):163-7. |
|
Dilated superficial veins (non-varicose) |
Palpable cord |
Dilated superficial veins are caused by obstruction of the deep venous system. Palpable cord refers to palpable superficial veins, which is a sign of superficial phlebitis. |
Clinical features of PE
- Hellenic J Cardiol. 2007 Mar-Apr;48(2):94-107.
- JAMA. 2003 Dec 3;290(21):2849-58.
PEs are frequently asymptomatic. Symptomatic patients most commonly present with dyspnea. Signs of DVT are only found in about 1/3 of PE patients.
| Symptoms | Corresponding sign(s) | Mechanism |
| Dyspnea* | Tachypnea*, decreased air entry, localized rales, wheezing | Hyperventilation to compensate for increased dead space and in response to chemical mediators from platelets.Dyspnea is a symptom of central, which causes more severe hemodynamic consequences because of occlusion of larger vessels. *Most common symptom and sign, respectively. |
| Parasternal heave, loud P2, increased JVP | Increased pulmonary pressure (from vasoconstriction) causes right ventricular overload (loud P2) and right ventricular dilatation (parasternal heave). Right-sided backward heart failure causes increased JVP, and eventually left-sided heart failure (tachycardia). | |
| Palpitations | Hemodynamic signs: Tachycardia | See above. Tachycardia is a sympathetic response to decreased cardiac output. |
| Pleuritic chest pain | Pleural friction rub, signs of pleural effusion (stony dullness on percussion, decreased fremitus) | PE near the pleura (peripheral PE) causes ischemia to the region, resulting in inflammation. Since the pleura is innervated, inflammation will produce localized pleuritic chest pain. Inflammation also increases the permeability of the pleural surface, leading to accumulation of exudative pleural fluid (pleural effusion). |
| Hemoptysis and cough | PE causes damage to the pulmonary vasculature, which leads to bleeding into the airways. Cough is usually nonproductive, and may be triggered by irritation of the pleura or the airways. Am J Med. 2007 Oct;120(10):871-9. |
|
| Syncope | Hypotension, cyanosis | Decreased left ventricular filling, causing forward heart failure. |
Diagnosis of DVT
- JAMA. 2006 Jan 11;295(2):199-207. (Discussion of Wells DVT score here)
- Chest. 2012 Feb;141(2 Suppl):e351S-418S. (2012 Chest Guidelines)
Diagnosis starts with history (risk factors) and physical, which can be used to generate a pretest probability using a validated clinical prediction rule, such as the Wells DVT score (see JAMA reference above). Patients with high likelihood of DVT can be further tested with compression ultrasonography, where the length of the proximal veins (popliteal and femoral) is sequentially compressed with the ultrasound probe. Normal veins are easily occluded with moderate external compression, but a DVT will prevent occlusion of the vein lumen. Ultrasonography is both sensitive and specific for DVTs.
- A D-dimer level can be done to rule-out DVT in individuals with low pretest probability (see discussion in Diagnosis of PE).
- Contrast venography is considered the gold standard for diagnosis of DVT, although this is rarely done because it is invasive, expensive, and not readily available. Contrast is injected into the dorsal foot vein, and the leg is imaged with CT scan or MRI.
Diagnosis of PE
JAMA. 2003 Dec 3;290(21):2849-58.
Diagnosis is based on history and physical, and confirmed with CT or V:Q scan if the clinical suspicion is high. The Wells criteria can be used to determine risk (pretest probability) of PE.
|
|
Criteria |
Points |
|
1 |
Clinical signs/symptoms of DVT |
3 |
|
2 |
No other diagnosis more likely than PE |
3 |
|
3 |
Tachycardia: heart rate > 100 |
1.5 |
|
4 |
Immobilization for > 3 days (e.g. strict bed rest) OR Surgery in the previous 4 weeks |
1.5 |
|
5 |
Previous DVT or PE |
1.5 |
|
6 |
Hemoptysis |
1 |
|
7 |
Malignancy |
1 |
|
|
Low risk (<2): 3% pretest probability Moderate (2-6): 20% High (>6): 63% Thromb Haemost. 2000 Mar;83(3):416-20. |
|
Note on D-dimer: In low-risk patients with symptoms that suggest PE, a D-dimer can be used to rule out PE if negative (high sensitivity, low specificity). D-dimer level is measured in the blood. As explained above, it is a degradation product of fibrin, which is elevated if a coagulation and fibrinolysis reaction happens in the body. In PE, endogenous fibrinolytic mechanisms try to dissolve the clot, which is the basis of an elevated D-dimer. However, the D-dimer level not specific and is elevated in any type of inflammatory process. Its clinical utility is limited to ruling out PE in those with a low pretest probability.
Treatment
Chest. 2012 Feb;141(2 Suppl):e419S-94S.
The goals of treatment for VTE are (i) anticoagulation to prevent further clot generation and (ii) thrombolysis if the thrombus is large enough to cause hemodynamic compromise.
Anticoagulation: Reduces further clot formation
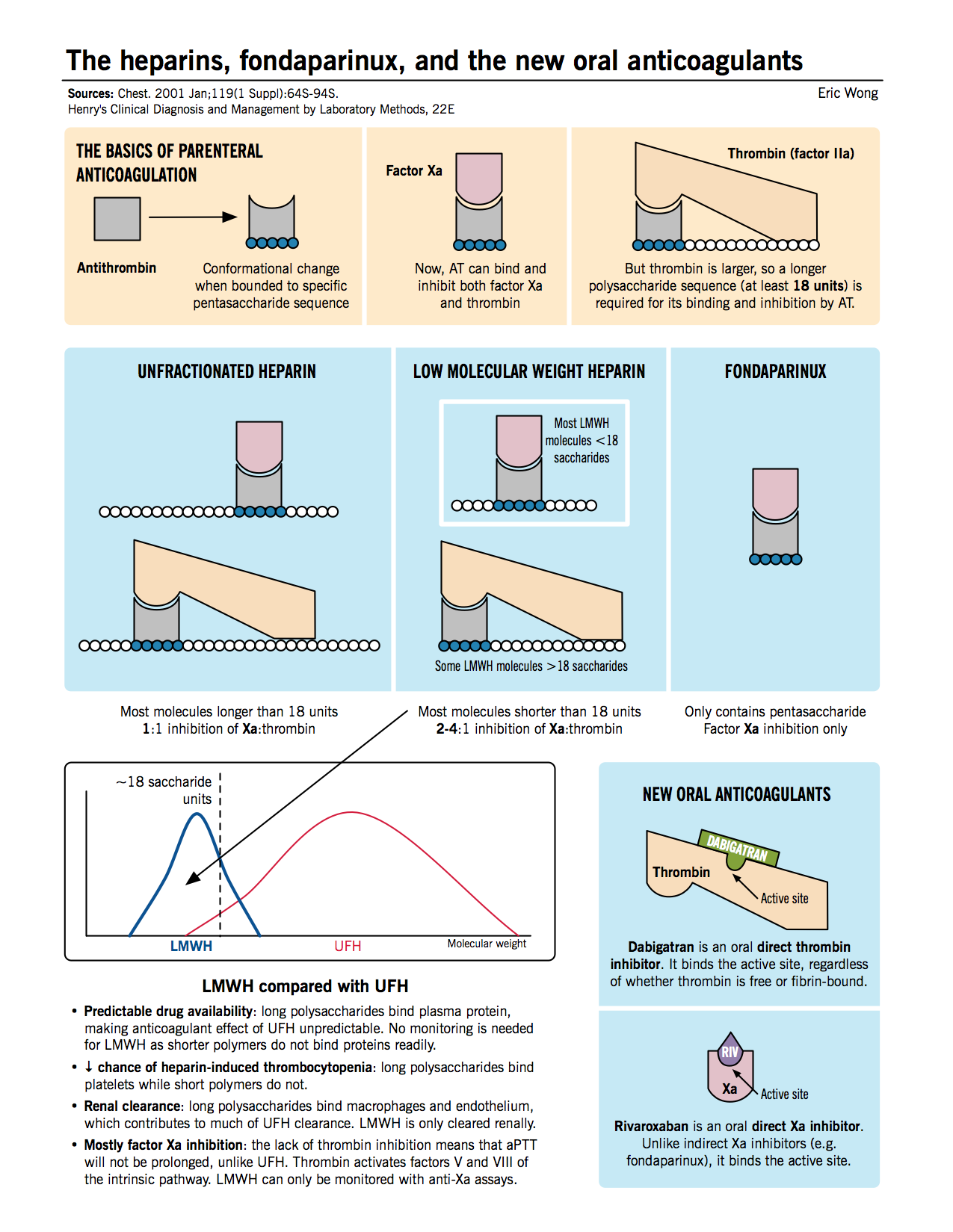
Anticoagulation with parenteral (intravenous or subcutaneous) and oral anticoagulants is the mainstay of VTE therapy. Typically, one of the parenteral agents (e.g. heparin, LMWH, or fondaparinux) or a new oral anticoagulant (e.g. rivaroxaban) is started first. The patient may be transitioned to a traditional oral anticoagulant (e.g. warfarin) for chronic anticoagulation.
- Unfractionated heparin (UFH): Inhibits the function of thrombin as well as Xa by inducing conformational changes in antithrombin, allowing it to bind the enzymes better.
- Low molecular weight heparin (LMWH): Functions similar to UFH, but due to the smaller average heparin chain length, accelerates the bridging of AT with Xa only, and not thrombin.
- Fondaparinux: A pentasaccharide sequence that directly binds to AT (at an allosteric site) and induces a conformational change allowing it to bind and inhibit factor Xa only.
- Rivaroxaban: A new oral anticoagulant that inhibits factor Xa by binding to its active site.
Chronic anticoagulation: For prophylaxis against future VTE
Any of the agents for acute anticoagulation can be used for chronic anticoagulation, but they are less convenient for outpatients due to the need for daily injections. Oral anticoagulation drugs are the mainstay for outpatient anticoagulation. Vitamin K antagonists (e.g. warfarin) were traditionally used, but newer agents, such as dabigatran and rivaroxaban, can also be used. In addition, aspirin is an antiplatelet agent that has been shown to reduce VTE events in recent trials.
- Vitamin K antagonists (e.g. warfarin): Warfarin inhibits the vitamin K dependent synthesis of calcium dependent clotting factors (II, VII, IX and X). Furthermore, warfarin also inhibits PS and PC (part of the endogenous anticoagulation pathway).
- The inhibition of PC and PS occurs faster than the other clotting factors, making warfarin acutely a procoagulant. Therefore, warfarin must be given concomitantly with acute anticoagulants at first (a process known as “overlapping”) to (i) prevent acute procoagulant effect and (ii) allow time for inhibition of vitamin K dependent factors. Once the patient’s international normalized ratio (INR) is therapeutic (2-3), acute anticoagulants can be discontinued.
- Warfarin has been the mainstay of chronic VTE therapy for over 50 years, but there are several issues with its use: (i) increased bleeding risk, (ii) teratogenicity in pregnancy, (iii) interaction with many foods and drugs, and (iii) close monitoring required because anticoagulation effect is not reliably predictable by dosage. New antithrombotic medications have been developed that are potentially safer than warfarin.
- Direct thrombin inhibitors (e.g. dabigatran): Directly block thrombin function by blocking the active site. Dabigatran is equivalent to warfarin in both prevention of recurrent clots and bleeding risk in patients with acute VTE, but it does not require monitoring due to its predictable therapeutic effect (RE-COVER trial).
N Engl J Med. 2009 Dec 10;361(24):2342-52. - Direct Xa inhibitors (e.g. rivaroxaban): Directly inhibit the function of Xa by blocking the active site. Unlike warfarin and dabigatran, rivaroxaban does not require overlapping with heparins. Rivaroxaban is equivalent to warfarin in short- and long-term prevention of PE in symptomatic patients, but it does not require monitoring or overlapping, and has significantly lower bleeding risk compared to warfarin (EINSTEIN-PE trial).
N Engl J Med. 2012 Apr 5;366(14):1287-97.
- Direct thrombin inhibitors (e.g. dabigatran): Directly block thrombin function by blocking the active site. Dabigatran is equivalent to warfarin in both prevention of recurrent clots and bleeding risk in patients with acute VTE, but it does not require monitoring due to its predictable therapeutic effect (RE-COVER trial).
- Aspirin: Although this antiplatelet agent is classically used to prevent arterial thrombosis, new evidence suggests that it can also be used for recurrent VTE prevention. Daily aspirin (100mg/day used in trials) can reduce VTE recurrence by approximately 1/3. Aspirin, although not as effective as other anticoagulants, may be used if the patient is intolerant of anticoagulants.
N Engl J Med. 2012 Nov 22;367(21):2039-41.
Thrombolysis: Breaks down the thrombus
- Tissue plasminogen activator (tPA): activates plasminogen (Pg) to plasmin (Pn), which cleaves the thrombus, generating soluble D-dimer products.
Contraindications to anticoagulation
- Thrombectomy: If a large thrombus creates hemodynamic compromise, and there are contraindications to thrombolysis, the clot can be surgically removed or by interventional radiology.
- Inferior vena cava (IVC) filter: Temporary IVC filters can be placed to stop the movement of clots from the deep veins of the lower extremity from travelling to the pulmonary vasculature.