Definition and etiology
- Lancet. 2010 Dec 11;376(9757):2018-31
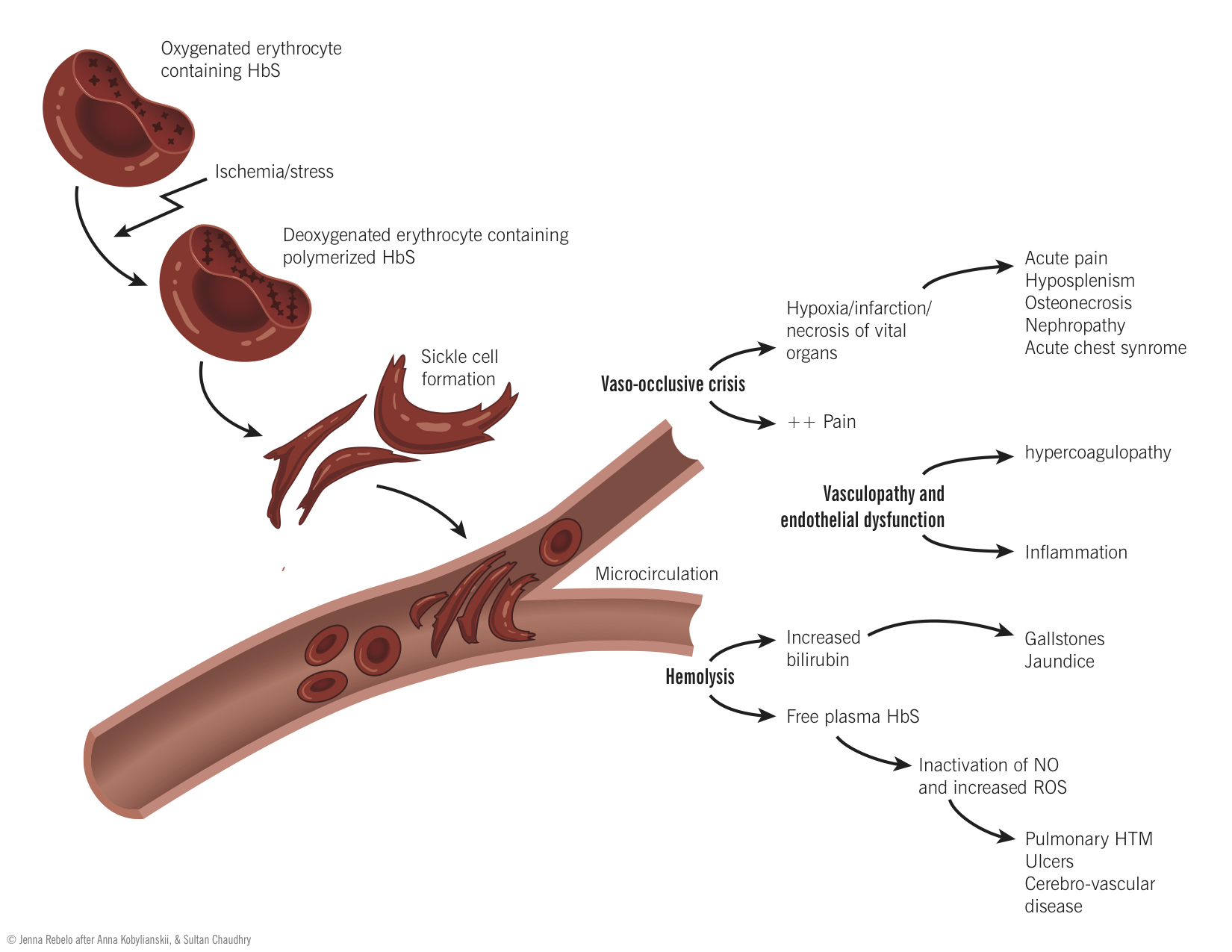
Sickle cell disease is an inherited, autosomal recessive, condition caused by several mutations in the β-globin gene. These mutations cause the sixth amino acid to be changed from glutamic acid to valine. The resultant hemoglobin (called HbS) has abnormal physiochemical properties, and is prone to polymerization with other hemoglobin molecules under conditions of low oxygen tension. This has a number of adverse affects on erythrocytes.
The normally freely flowing cytosol of red cells become viscous making the red cell much less deformable and impairing its ability to traverse tight capillary beds. As HbS continues to polymerize the entire RBC is deformed giving the characteristic sickle shape.
Pathophysiology of disease and complications
- Lancet. 2004 Oct 9-15;364(9442):1343-60
- Lancet Neurol. 2006 Jun;5(6):501-12.
- Int J Infect Dis. 2010 Jan;14(1):e2-e12.
- Semin Thromb Hemost. 2011 Apr;37(3):226-36
- Semin Hematol. 1997 Jul;34(3 Suppl 3):2-7.
| Complication | Mechanism |
| Vaso-occlusive crisis | Due to the deformed shape, HbS induces RBC membrane damage leading to calcium influx into the cell. Calcium influx leads to crosslinking of the membrane proteins and activating channels that allow for the efflux of potassium and water from the cell. This leads to RBC dehydration exacerbating the sickling.Vaso-occlusive crisis results from the sickle red cells obstructing and reducing blood flow to the vital organs leading to ischemia, necrosis and pain. Repeated episodes lead to bone infarction and necrosis; and bone marrow degeneration occurs overtime. Long bones are affected most commonly, but pain episodes can affect any bone marrow-containing structure, including the ribs, sternum, vertebral bodies, and skull.Pulmonary fat embolism can be a life threatening complication of bone marrow infarction in patients with SCD and precipitate Acute Chest Syndrome (ACS).
|
| Hemolysis | Sickle cells are mechanically weak and are prone to intravascular hemolysis. However, the more important mechanism leading to decreased red cell survival time is the extravascular hemolysis that occurs when inflexible cells are trapped in the spleen and phagocytosed by the reticuloendothelial systemBone marrow tries to compensate by increasing RBC production but it cannot match the rate of destruction.
Complications of increased hemolysis include cholelithiasis; due to excessive bilirubin production. |
| Hyposplenism and Infection | Encapsulated bacterial infections: Splenic sequestration of sickle cells leads to splenic congestion, as manifested by splenomegaly, and reduced immune function. Since the spleen is important for macrophage phagocytosis of encapsulated bacteria, patients with SCD are prone to bacteremia with pathogens like Streptococcus pneumoniae (S. pneumoniae), Haemophilus Influenzae (H. Influenzae) and Neisseria meningitidis (N. Meningitidis). These pathogens, normally causing localized disease, may cause life-threatening sepsis in patients with sickle cell disease.Furthermore, patients with SCD develop pneumonias, predominantly from atypical organisms, such as, Mycoplasma pneumoniae, Chlamydia pneumoniae and Legionella. Respiratory viruses are also common causes of pulmonary infection, while S. pneumoniae and H. influenza type b are uncommon.Osteomyelitis and septic arthritis can affect minority of patients with SCD due to both, bone damage and poor splenic function. The leading pathogens include Salmonella, S. Aureus and other gram negative bacteria.
Autosplenectomy: Continued splenic dysfunction eventually leads to infarction and loss of splenic function, which is referred to as autosplenectomy or functional asplenia. Dysfunctional complement system has been proposed to contribute to the infectious complications in SCD, however, this has yet to be proven. |
| Pulmonary and Cardiovascular complicationsLancet. 2000. April; 355(9214):1476-78 | Neurologic complications are related to vasoocculusive crisis and include strokes and silent strokes.The acute pulmonary complications of SCD, collectively referred to as the acute chest syndrome (ACS). It is defined as the appearance of new infiltrate with pulmonary symptoms, presence of fever, hypoxia and chest pain (minority of patients might not have an infiltrate initially). Precipitants of ACS include infections (see above for details), infarction and/or pulmonary fat embolism.No specific cardiomyopathies are present however, two cardiac complications occur; chamber enlargement related to significantly increased forward output due to chronic anemia and sleep/wake oxygen desaturations and secondly, myocardial infarction from vasoocclusion of the coronary arteries. Rarely, RV dysfunction (Cor pulmonale) may develop due to Pulmonary arterial hypertension. |
Treatment
- Haematologica. 2004 Mar;89(3):348-56.
- Lancet Neurol. 2006 Jun;5(6):501-12.
- Curr Opin Pediatr. 2009 Feb;21(1):15-21.
- BMJ. 2003 November 15; 327(7424): 1151–1155.
- Vaso-occlusive Crisis: Vaso-occlusive crisis is managed with rehydration therapy to reverse the dehydration and hypoxia. Rehydration allows for increased RBC water and electrolyte uptake (see above for RBC water loss during sickle cell crisis).
- Transfusions/Erythrocytapheresis: Transfusions and replacement of sickle cells with those of normal RBCs allows to keep HbS concentrations <30%. This is of particular importance during operative procedures (high risk for precipitating crisis).
- Pain: The major factor affecting quality of life in SCD is pain. Hydroxyurea, an anti-neoplastic drug increases production of fetal hemoglobin, is used to prevent pain. Fetal hemoglobin carries 2 alpha and 2 gamma globin chains. Since the mutation is in the beta globin, upregulation of gamma globin chains reduces the pain associated with vaso-occlusive crisis.
- Acutely painful episodes are managed with morphine.
- Chronic pain is also managed using NSAIDs
- Infections: Infections are managed using antibiotic prophylaxis and early immunization for Pneumococcus and Meningococcus infections.
- Coverage for atypical organisms in ACS is necessary.
- Gallstones: Management is different from that of general population (see GI section).
- Priapism: Oral hydration therapy to hydrate RBCs and oral anelgesics to manage pain.
- Stroke prevention: Guidelines for general stroke population apply to patients with SCD. Regular transfusions have shown to be of significant benefit in stroke prevention, most likely due to reduction of sickle cells.
- Chronic Anemia: Due to the high turnover of RBCs, chronic anemia is not a major issue
- Hematopoietic Stem Cell Transplant (HSCT): Patients with severe disease refractory to current standard therapy (hydroxyurea), under the age of 17, or those with prior SCD related organ damage (eg, stroke, acute chest syndrome, frequent painful episodes, multiple sites of osteonecrosis) can be considered for HSCT.
- Myeloablative and non-myeloablative approaches have been studied. Majority of the data support a myeloablative HSCT, whereas non-myeloablative HSCT still remains under investigation.